Modos de acción de los oncogenes en tumores humanos asociados
FACTORES DE CRECIMIENTO
Los factores de crecimiento estimulan a células normales a
proliferarse. Las células cancerosas adquieren también la
capacidad de producir factores de crecimiento, lo que genera un ciclo
autocrino.
El ciclo autocrino es un elemento importante en la
patogenia de varios tumores, la mayoría de los casos no está alterado ni mutado
el propio gen de factores de crecimiento. Los productos de otros oncogenes que
se sitúan a los largo de muchas vías de transducción causan una sobreexpresión
de los genes de factores de crecimiento, esto genera que las células sinteticen
grandes cantidades de dichos factores.
La proliferación que estimula los factores de crecimiento contribuye al
fenotipo maligno mediante un incremento de riesgo de mutaciones espontáneas o
inducidas en la población celular en proliferación.

A continuación, se muestra una tabla en donde se se explica la
clasificación de estos factores de crecimiento y los tumores que se ven
asociados.

En 1948, se aisló y se caracterizó el factor de crecimiento
derivado de plaquetas tipo B (PDFG-B), y como resultado del análisis de su
secuencia se mostró que era idéntica a la del oncogén V-SIS. El oncogén SIS fue
el primer oncogén que fue asociado a las funciones de las células normales. Las
células que expresan el oncogén SIS son transformadas por medio de un mecanismo
de estimulación autocrino por parte del PDGF-B, ya que la misma célula que
expresa el oncogén también presentan receptores para PDFG-B en la superficie.
Este mecanismo produce una autoestimulación constitutiva que puede llevar al
crecimiento maligno de las células.
En fibroblastomas y glioblastomas se
ha observado sobreexposición de PDGF-B. En los mesoteliomas se
encuentra sobreexposición de PDGF-A, el cual también es
habitualmente expresado en las células normales.
Otros factores de crecimiento con capacidad oncogénica son los de la
familia de los factores de crecimiento de fibroblastos (FGF), entre
los que se ha localizado tres formas oncogénicas: int-2, hst y fgf-g.
Una elevada expresión de TNF, el cual es miembro de la familia del
factor de crecimiento epidérmico (EGF), se ha encontrado en ciertos carcinomas.
El factor de crecimiento wnt-1 y otros miembros de su familia,
se han relacionado con retinoblastoma, cáncer de estómago, fibroadenomas, entre
otros. (1)
RECEPTORES DEL FACTOR DE CRECIMIENTO
Los receptores del factor de crecimiento en las células
normales y en las cancerígenas son prácticamente las mismas, ambas son
transmembrana con dominios externos de unión al ligando y dominios internos
citoplasmáticos con unión a tirosina cinasa; con la única diferencia que el
receptor transmembrana codificado a partir de un oncogén no necesita del factor
de crecimiento para realizar sus funciones a partir de la tirosina cinasa. Es
decir, este receptor transmembrana normalmente se activaría momentáneamente
debido al estímulo del factor de crecimiento seguida de la dimerización y
consecuente fosforilación con tirosina de varios sustratos que forman parte de
la cascada de señalización. Por otra parte los receptores mutados u oncógenas
de estos receptores se asocian a la dimerización y activación sin unión al
factor de crecimiento.
Los receptores pueden activarse por mutaciones,
redistribución genética o sobreexpresión; por ejemplo el protooncogén RET es un
receptor de tirosina cinasa para el factor neutrófico derivado de la línea
celular glial durante el desarrollo neural que puede mutar, causando así
que la proteína RET que se codifica a través de ella, se asocie a las
neoplasias endocrinas múltiples (NEM) tipo 2A y 2 B, así como también está
presente en el carcinoma medular de tiroides familiar. El NEM tipo 2A causa
dimerización y activación constitutivas conduciendo a carcinomas medulares de
la tiroides y tumores suprarrenales y paratiroideos. El NEM 2B por su parte
altera el sustrato especifico de la tirosina cinasa y conducen tumores
tiroideos y suprarrenales sin alteración de las paratiroides.
Así también se han encontrado mutaciones en genes que
codifican diferentes receptores, como en la leucemia, el FLT3 que codifica el
receptor de tirosina cinasa similar al FMS, en esta el receptor PDGF está unido
a un segmento de transcripción de ETS, causando una dimerización permanente de
PDGF.
PROTEÍNAS IMPLICADAS EN LA TRANSDUCCIÓN DE LA SEÑAL
Se han encontrado varios ejemplos de oncoproteínas que
imitan la función de las proteínas transductoras de señal citoplasmáticas
normales. En su mayoría, estas proteínas se localizan estratégicamente en la
capa interna de la membrana plasmática, donde reciben señales del exterior de
la célula y las transmiten al núcleo de la célula.

La oncoproteína transductora de señal mejor estudiada es la
familia RAS de proteínas que se unen a la guanosina trifosfato (proteínas G).
El oncogén RAS: los genes RAS, de los cuales existen tres
(HRAS, KRAS, NRAS), la mutación puntual de estos genes es la anomalía aislada
más frecuente de los protooncogenes en tumores humanos.
RAS tiene un importante papel en las cascadas de señales a
favor de corriente de los receptores de factor de crecimiento, dando lugar a
mitosis. El ciclo ordenado de la proteína RAS depende de dos reacciones: 1)
intercambio de nucleótidos, que activa la proteína RAS, y 2) hidrolisis de GTP,
que convierte el RAS activo, unido a GTP, en la forma inactiva, unida a GDP.
Se han identificado varias mutaciones puntuales de RAS
diferentes en las células cancerosas. Los residuos afectados se sitúan en el
bolsillo de unión de GTP o bien en la región enzimática esencial para la
hidrolisis de GTP, y por ello reducen considerablemente la actividad GTPasa de
la proteína del RAS.
Alteraciones de las tirosina cinasas sin receptor: en
algunos casos las mutaciones se deben a translocaciones o reordenamiento
cromosómicos que dan lugar a genes de fusión que codifican tirosina cinasas
activas de forma constitutiva. Un ejemplo importante de este mecanismo oncógeno
implica la tirosina cinasa c-ABL.
TABLA NO. 1
PROTEÍNAS INVOLUCRADAS EN LA TRADUCCIÓN DE LA SEÑAL.

PROTEINAS REGULADORAS NUCLEARES
La p53 es una de las principales proteínas relacionadas
con los mecanismos de regulación de “puntos control” o check-points del
ciclo celular, que centraliza la coordinación de otros procesos relacionados
con el daño celular como son la reparación de daños en las bases o roturas
dobles de cadena, la progresión del ciclo celular y la muerte por apoptosis. Es
comúnmente denominado el “guardián del genoma”, porque mantiene la
estabilidad genética celular. Su relación con el cáncer es bien conocida por la
alta frecuencia de alteraciones observadas, como por la frecuencia de aparición
de tumores en personas con mutaciones germinales de p53 (Síndrome
Li-fraumeni). Además, en tumores que no muestran mutaciones del gen, la
función de la proteína puede estar alterada debido a su secuestro
citoplasmático por oncoproteínas virales.
La proteína Bcl-2, es también un importante regulador de
control y su gen codificador está en el cromosoma 18. En condiciones normales,
la expresión de Bcl-2 disminuye cuando las células están maduras o cuando
tienen que ser eliminadas, mientras que se expresa en células que deben
sobrevivir, como las células hematopoyéticas precursoras y las células del
sistema nervioso.Bcl-2 tiene un importante papel en la embriogénesis, donde la
mayoría de las células expresan altos niveles de Bcl-2. En general, las células
que la expresan bloquean la apoptosis por lo que promueven la
supervivencia celular, facilitando la adquisición de mutaciones y la
transformación maligna.
Bax (Bcl-2 associated X protein) es una proteína
cuyo gen codificador está en el cromosoma 19. Aunque posee una alta homología
con la proteína Bcl-2 carece del dominio BH4. Participa en la ruta de apoptosis
mitocondrial induciendo la liberación del citocromo C. Se ha observado que Bax se asocia con el complejo del
poro de la mitocondria (PT) que participa en la regulación del Ca2+ de
la matriz, ph, potencial de membrana mitocondrial, etc. La proteína Bax se une
a un componente de este complejo induciendo la apertura del poro con la
consiguiente rotura del potencial de membrana mitocondrial y liberación de moléculas
pro-apoptóticas como el citocromo C.

REGULADORES DEL CICLO CELULAR
¿Qué es
el ciclo celular?
El ciclo celular es un proceso
que consiste en la división de una célula para crear una copia exacta de sí
misma, permitiendo crecer y así reemplazar a las células a medida que se
desgastan. En los animales, el ciclo normalmente se completa en alrededor de 24
horas para los distintos tipos celulares, aunque algunas, como las de la piel o
las tumorales se mantienen en división constantemente.
La mitosis comienza con células en reposo
(Fase G0), las cuales deben de ser estimuladas por factores de crecimiento para
lograr entrar en el ciclo, esto da inicio al período de crecimiento (Fase G1)
en el que la célula se prepara para un período de síntesis de ADN (Fase S). Cerca
del final de G1, existe un punto de restricción (R), en que el ADN es reparado
en caso este dañado, de no ser así el ciclo sigue en adelante. Una vez los
cromosomas han sido duplicados, la célula entra a un segundo período de
crecimiento (Fase G2), cuando se prepara para dividirse en dos células hijas
durante el periodo de mitosis (Fase M). Esta fase se divide en una serie de
pasos que comienzan con la profase, luego metafase, anafase, telofase y por
último, citocinesis, que divide la célula en dos iguales.

CELL CYCLE
Regulación
del ciclo celular y el cáncer
El proceso del ciclo celular está controlado
por diversos factores de crecimiento, que se encargan de determinar el
comportamiento de la célula, desde la decisión de crecer, diferenciarse o morir
por apoptosis. Todos los factores realizan su mecanismo de regulación a través
de ciertas proteínas que pueden ser positivas o negativas.
Entre las proteínas regulatorias con mayor
importancia podemos encontrar las ciclinas, que conforman la subunidad
regulatoria de otras proteínas conocidas como proteínas cinasas dependientes de
ciclinas (CDK). Además de las ciclinas, existe
un grupo de genes conocidos como de respuesta temprana, cuyo papel es muy
importante en las fases tempranas del ciclo celular, y que al igual que las
anteriores son activados por los factores de crecimiento. Los ARN mensajeros
tempranos incluyen c-fos, c-jun y c-myc, los cuales aparecen pocos minutos
después de la estimulación mitogénica. Se ha demostrado que la inducción del
ARNm del gene c-myc es necesaria y suficiente para la transición de la fase G1
a S. Por otro lado, existen proteínas cuya acción principal es la supresión
tumoral como la proveniente del gen del retinoblastoma RB y la familia de las
proteínas p53, que actúan regulando la fase G1.
A
continuación se explicaran brevemente los principales reguladores del ciclo
celular.
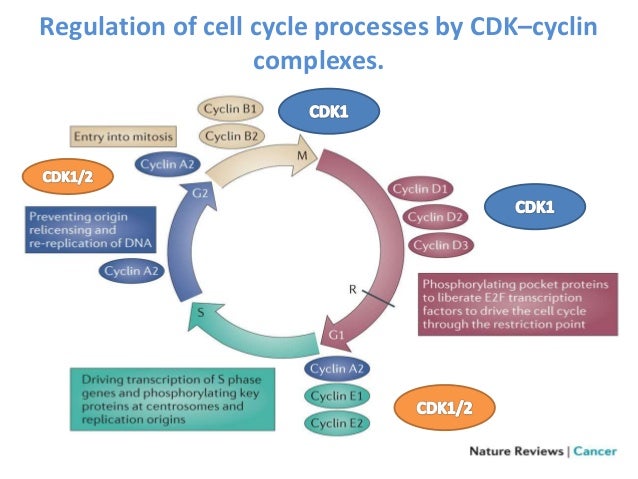
Ciclinas y Cdk
La progresión del ciclo celular se activa
de manera directa por una serie de heterodímeros formados por las ciclinas y
las cinasas dependientes de ciclinas (CDKs).
La ciclina D actúa como un sensor de
crecimiento y provee una unión entre la estimulación de la mitosis y el ciclo
celular. La decisión de una célula para entrar en fase S está estrechamente
controlada por la ciclina D1 que se une a la CDK4 y CKD6 en la fase G1 y la
ciclina E unida a CDK2, seguido del complejo ciclina A-CDK2 a lo largo de la
fase S. La ciclina D, al formar el complejo con las CDK, activa la acción de la
cinasa cuyo sustrato principal es la proteína retinoblastoma (Rb). Las
aberraciones en la expresión de la ciclina D1 han sido registradas en diversos
canceres humanos. Por ejemplo, en muchos tumores de mama y en el desarrollo de
la mama durante el embarazo, se da una sobreexpresión de la ciclina D. La ciclina
D2 y la D3 también presentan una sobreexpresión en casos de cáncer de colon y
mama y en leucemias mieloides agudas. Tanto la ciclina A como la E se sobre expresa
en el carcinoma de pulmón.
Los
reguladores negativos tales como la RB y los CDKI actúan como una barrera
energética potencial en los complejos ciclinas-CDK para inducir la entrada al
ciclo celular. Cuando estas barreras son removidas debido a una mutación, se
reduce la actividad cinasa de las CDK que es necesaria para entrar al ciclo, dándose
una regulación negativa. Cuando las células que no están proliferando regresan
a un estado proliferativo o se incrementan los niveles de ciclinas y/o se
disminuyen los CDKI o la función de la RB, las células son capaces de entrar a
un estado maligno por la alteración de sus inhibidores.
Proteína rb
La proteína el retinoblastoma (RB) se
encarga de la supresión tumoral al inhibir la proliferación celular promoviendo
la senescencia y la diferenciación. La RB es fosforilada después de un estímulo
mitogénico, promovido por la unión de ciclina D-CDK, pero se degrada en respuesta
a un estímulo de muerte. En el estado hipofosforilado, la proteína supresora de
tumores Rb, está activa y lleva a cabo su función mediante la inhibición de la
progresión del ciclo celular, bloqueando a los factores de transcripción E2F1,
E2F2 y E2F3a, que son esenciales para la expresión de genes que le darán
continuidad al ciclo. En el momento en que se fosforila, el gen del retinoblastoma
(pRb) libera a E2F, lo cual lleva a la trascripción de genes críticos para la
progresión de células de la fase G1 a la fase S del ciclo celular.
Se cree que la alteración en la función de
Rb se puede deber a una mutación del gen p16. El gen p16 se encuentra
relacionado con el ciclo celular. Su función es la inhibición de las cinasas
dependientes de ciclinas 4 y 6, lo cual evita la fosforilación de la pRb e
impide la progresión del ciclo celular desde la fase G1 a la fase S.

P53
El sistema de vigilancia de la proteína
p53 se encuentra constantemente comprobando el rendimiento óptimo de todos los
procesos del ciclo celular y particularmente aquellos relacionados con la
síntesis de ADN. Su incremento inhibe la progresión de G1 a S en las células
con ADN dañado, lo que previene la acumulación de este ADN en las siguientes
generaciones. Este sistema es sensible tanto al estrés celular proveniente de fuentes
endógenas como exógenas, que a menudo resulta en el daño genético. Si este daño
no es demasiado grave, la p53 detiene el ciclo celular, mientras el daño es
reparado. Sin embargo, si el daño es demasiado grave, la célula es impulsada
hacia la senescencia o la apoptosis inducida por p53, esto debido a su
interacción directa con la endonucleasa AP y la ADN polimerasa que están
implicados en la reparación por escisión. La p53 también induce proteínas como GADD45
que colaboran en la reparación del ADN. Si el daño se repara correctamente, p53
estimula la síntesis de Mdm2, activando su autodestrucción y la progresión en
el ciclo celular. Si el daño no puede ser reparado, la célula puede entrar en
apoptosis o en senescencia, ambos inducidos por p53.
La pérdida de la función de la p53 por
mutación o inactivación produce inestabilidad genómica, apoptosis débil y
restricción del ciclo celular. La alteración de la p53 es la mutación más común
en el cáncer humano. Alrededor de la mitad de todas las malignidades humanas,
incluyendo muchos cánceres urológicos, tienen mutaciones en la p53.

REGULACIÓN DEL CICLO CELULAR
PERDIDA DE LA REGULACIÓN DEL CICLO CELULAR Y CANCER
Referencias Bibliográficas:
1.
Centro de investigación del cáncer. Factores de
crecimiento. [en línea] 2017. Universidad de Salamanca:
España. [Citado 15 de agosto 2017]. Disponible
en: http://www.cicancer.org/es/factores-de-crecimiento
2. Vermeulen K, Dirk R, Bockstaele V, Berneman Z. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. [en línea] 2003. Cell Prolif: Estados Unidos. [Citado 15 de agosto 2017] 36. 131-149. Disponible en:http://www.bath.ac.uk/bio-sci/hejmadi/cell%20cycle%20%26%20cancer%20rev.pdf
3. Laguna Cruz M, Valle Mendiola A, Soto Cruz I. Ciclo celular: mecanismos de regulación. [en línea] 2014.Rev. Vertientes:México. [Citado 15 de agosto 2017] 17 (2): 98-107. Disponible en:http://www.medigraphic.com/pdfs/vertientes/vre-2014/vre142e.pdf
4. López Marure R. La regulación del ciclo celular y el cáncer. México:Rev. Vertientes. 2003. 6(1): 40-44.

Comentarios
Publicar un comentario